8 min to read
Studying the COVID-19 genome sequences
This notebook is an attempt to answering some of the questions of bio-informatics in regards to the recent panademic outbreak of Corona virus, from a non-medical background person’s perspective. Every analysis presented here is based on the stuff I learnt and understood (or felt that I actually understood) about the genomes from the internet.
Some Context
[Ques.] What is Genome?
Genomes are considered to be genetic material of any organism. It consists of DNA(or RNA) which in turn can be considered as the blueprints of any organisms’ origin.
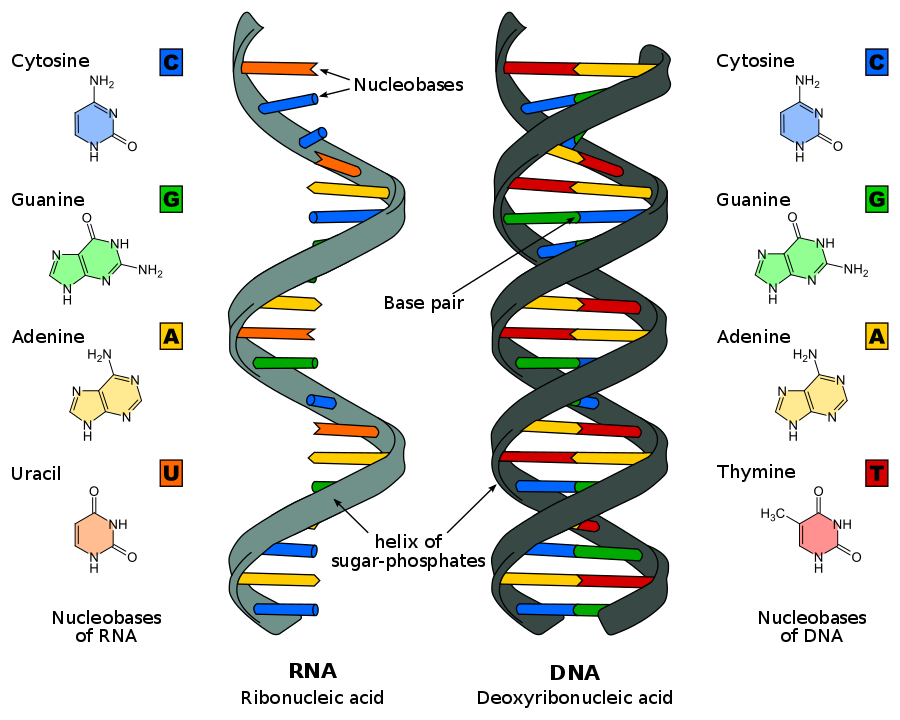
[Ques.] How to study Genomes?
A genome consists of a sequence of nucleotides that together make up all the chromosomes of any organism. These nucleotides are the basic building blocks of DNA and RNA. A DNA genome has 4 types of base nucleotides -
Adenine also represented as A,Thymine represented as T,Guanine represented as G andCytosine represented as C.
In RNA genome, the Thymine nucleotide is replaced with what is known as Uracil represented as U.
In short, any genome sequence is basically a combination of all these nucleotide in smoe order.
[Ques.] Can one find a cure for COVID-19 by just studying the genomes?
Not sure, as the molecular biology has not really been my field of study (never liked Biology tbh) but also because its a very vast subject where to confirm on a single hypothesis also takes months/years of evaluation and testing. Having said that, the whole purpose of this analysis is to help the ones who actually understand molecular biology get some data-driven insights to be able to preapare a RNA vaccine at the earliest.
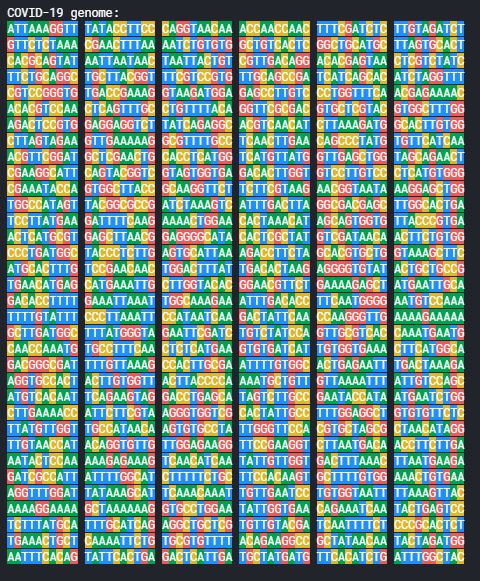
lets have a look at a snippet of one of the genomes,

having a huge sequence and with everyting in single color could be difficult to read. So let’s color-code these nucleotides in the sequences and have them represented as they are normally written.
Comparison of composition of genomes
Comparing the genomic composition, helps Vaccinologist do reverse vaccinology to discover candidate antigents for vaccine development by analyzing the genome of a pathogen or a family of pathogens.
For the comparison of composition of these genomes, I’ll be looking at the
- Oligonucleotide composition
- GC content
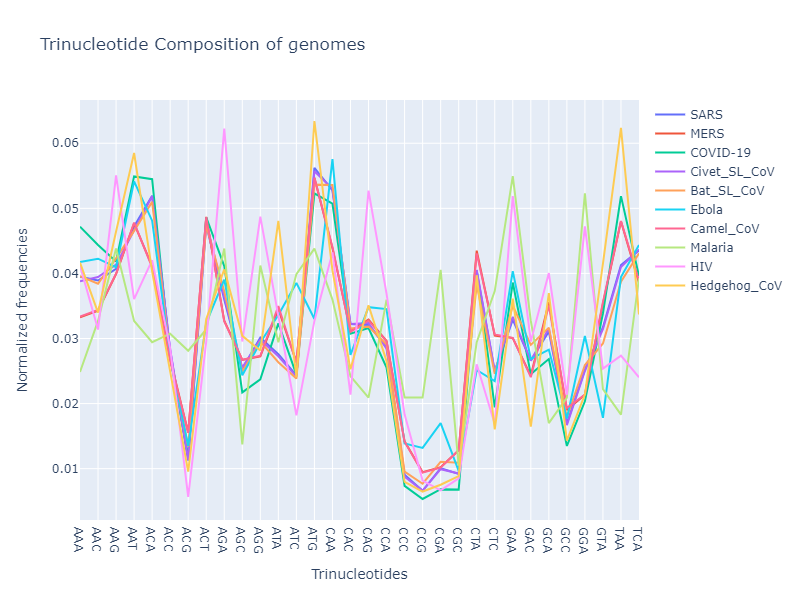
1. Oligonucleotide composition:
Oligonucleotides are DNA or RNA molecules made up of a sequence of nucleotides. The length of the oligonucleotide is represented as “k-mer” ($k$ being the number of nucleotides in one oligonulceotide). In this analysis, I’ll be looking at the 3-mers (or trinucleotides) and 4-mer (or tetranucleotides) compositions against their normalized frquency of occurrences.
Now lets have a look at the tetramer composition of these genomes
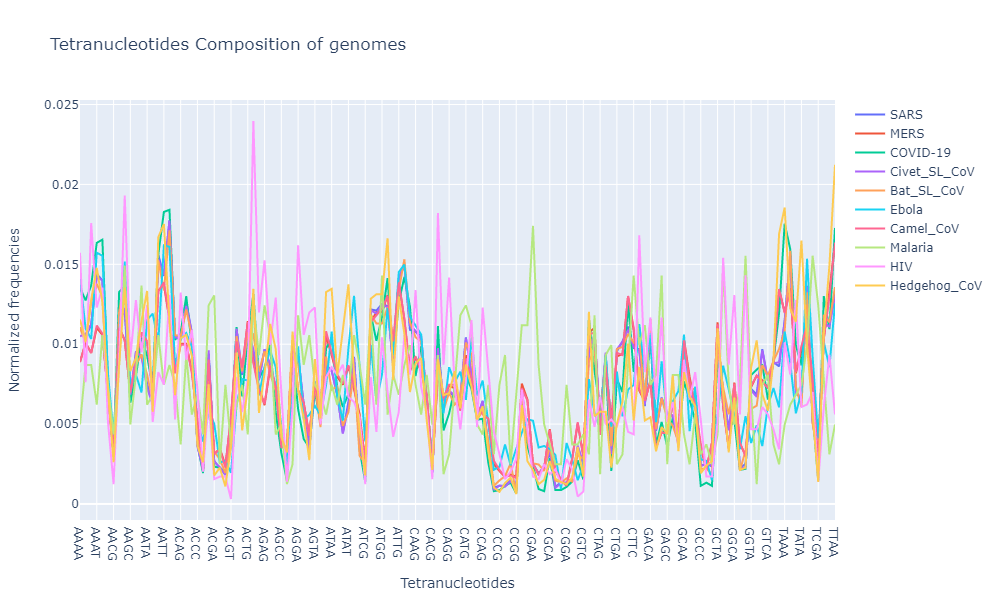
Well that’s alot to put in one plot, but the good thing with plotly is that you can always turn off a few legends to compare the values just among the selected few.
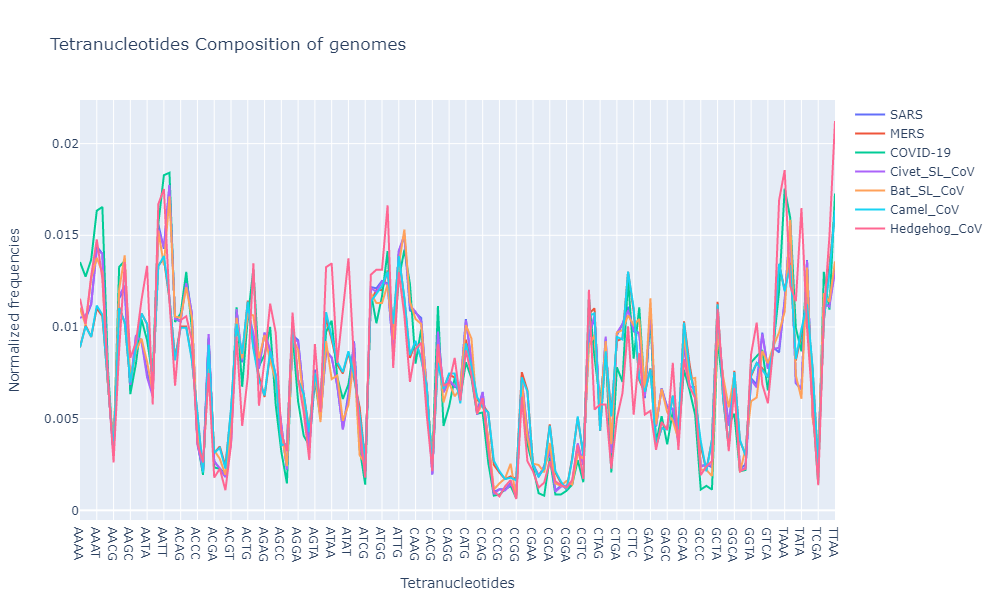
For a moment, lets just look at the tetranucleotide composition of all the “corona-virus infected species”.
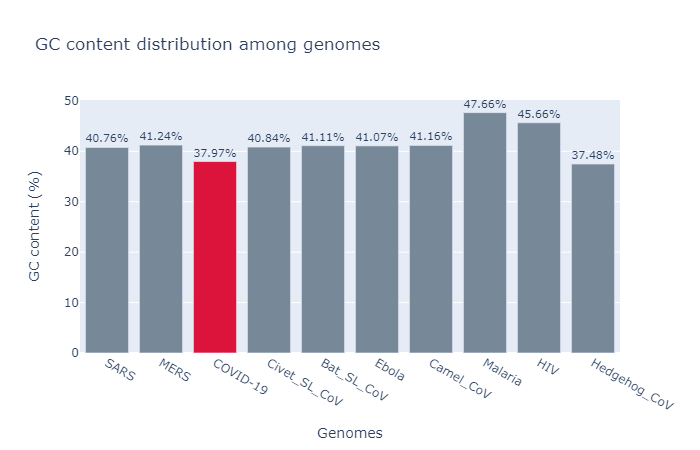
2. GC Content:
GC content (or Guanine-Cytosine content) is the percentage of nucleotides bases in a DNA/RNA molecule that are either guanine (G) or cytosine (C). GC content is always expressed as a percentage value and is supposed to remaine the same among genomes of same specie.
\(\frac{G + C}{A + C + G + T} \times 100\ \%\)
Proteins & Amino Acids
The most commonly occuring amino acids are abbreviated by using 20 letters from the english alphabet (all letters except for B,J,O,U,X and Z). The protein strings are constructed from these 20 alphabets as symbols. Henceforth, a Protein Sequence can also be incorporated using the nucleotide sequences.

The DNA codon table on wiki dictates the details regarding the encoding of specific codons into amino acids alphabets.
lets have a look at the protein sequences of a few of these genome before we go for a comparative analysis

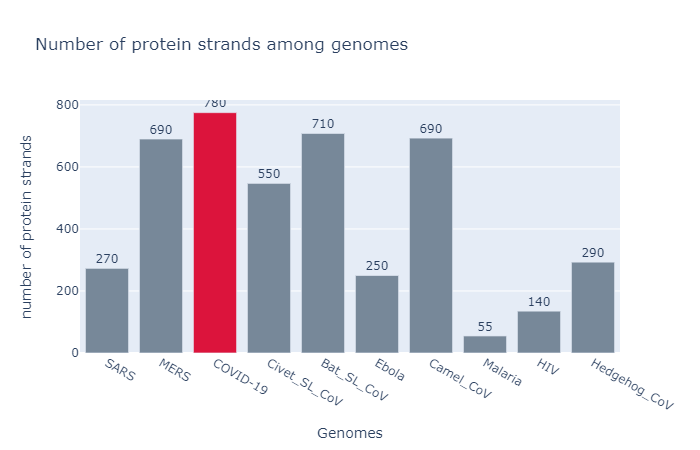
1. Protein Strands by genome
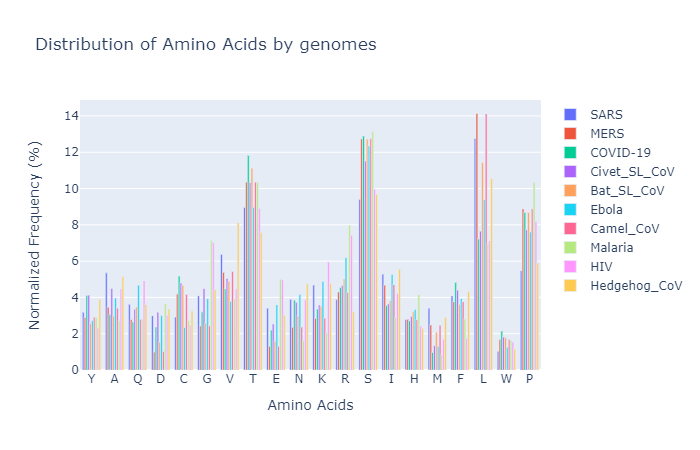
2. Amino Acids Distribution
3. Finding the ORFs (Open Reading Frames)
The stretch of codons from start-codon (ATG) to stop-codon (TAA/TAG/TGA) is called Open Reading Frame (ORF)
the highlighted sequences below represent the ORFs
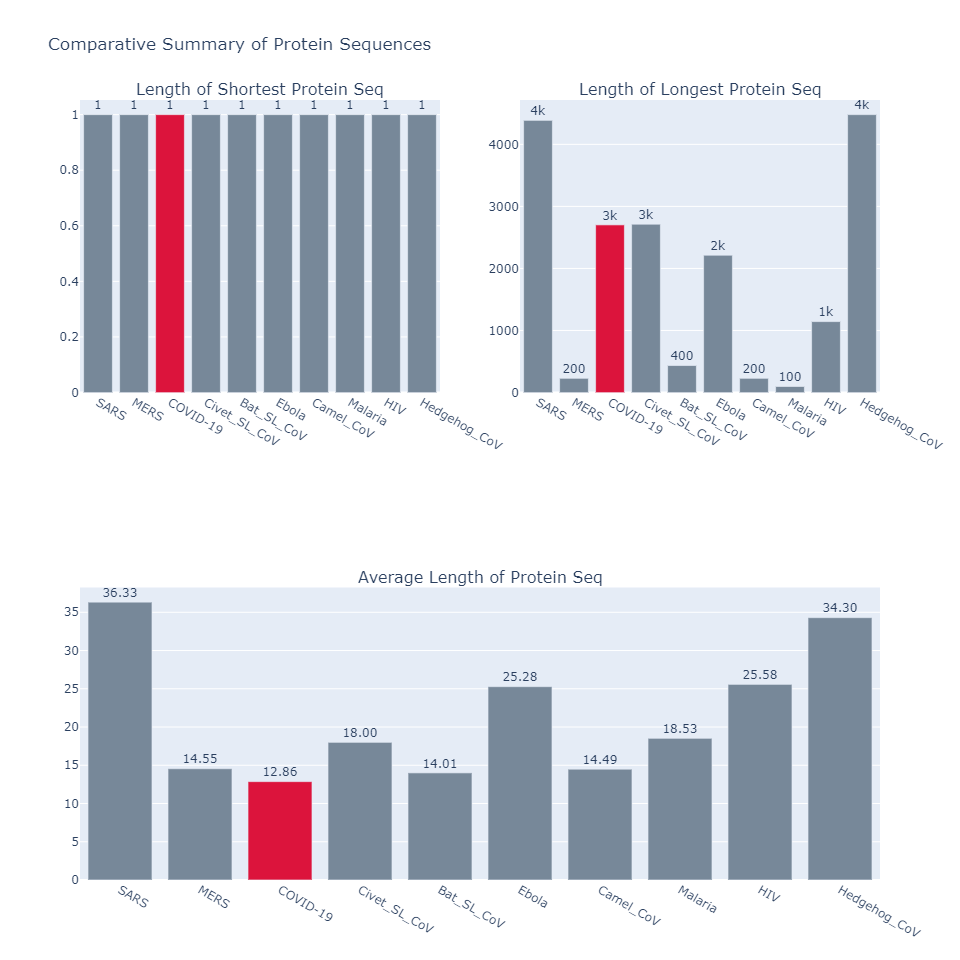
4. Comparative Summary of Protein Sequences
Sequence Alignement
The sequence alignment is a way of arranging the sequences of DNA/RNA or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. These in turn help in identifying the sequence similarities and developing homology models of protein structures. Alignments are often assumed to reflect a degree of evolutionary change between sequences descended from a common ancestor.
as the genomes sequences are too big in length, lets align and have a look at just the first few nucleodtides in the sequences
let us also have a look at the first few aligned protein sequences of these genomes

Pairwise Sequence Alignment
earlier we had aligned every sequence based on the Clustal algorithm, now let’s align every other genome with the covid19 genomes.
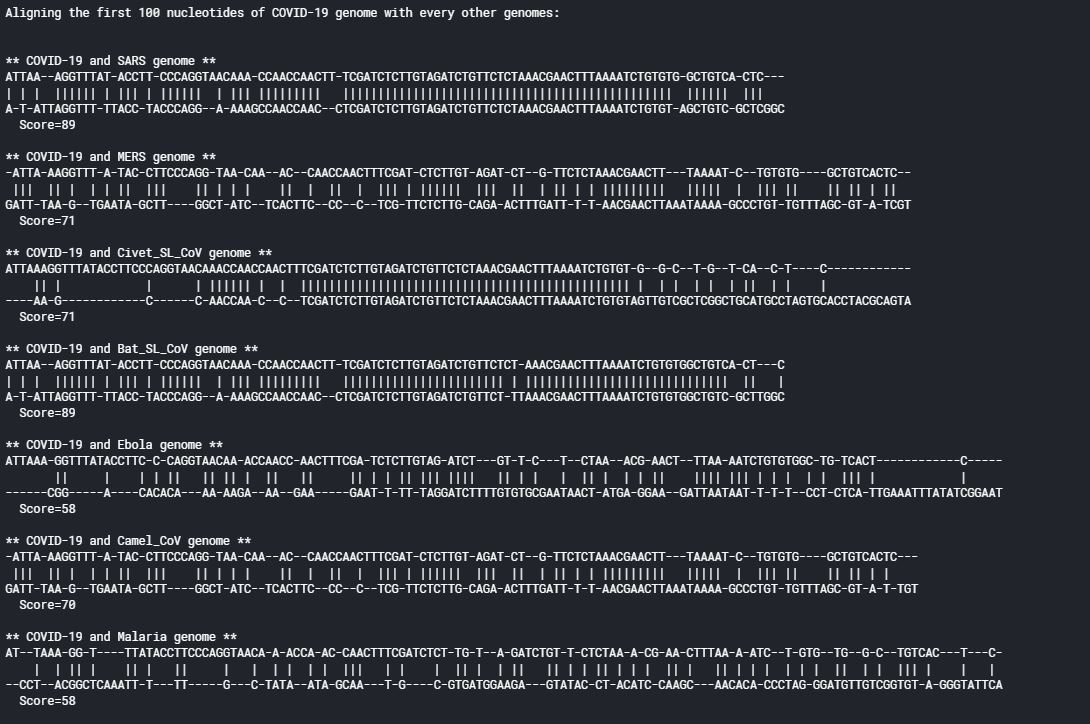
Genome Sequence Similarity
using Clustal algorithm
for establishing the similarity among genomes, we’ll be changing the scoring parameter
for every match there will be a +1 point
for every mismatch there will be a -1 point
for every open gap there will be a -0.5 points
for every extended gap there will be a -0.1 point
Similarity scores between
COVID-19 & SARS genome sequences: 20885.00000000038 (69.84%)
COVID-19 & MERS genome sequences: 15122.599999998136 (50.57%)
COVID-19 & Civet_SL_CoV genome sequences: 20616.90000000066 (68.95%)
COVID-19 & Bat_SL_CoV genome sequences: 20706.000000000255 (69.24%)
COVID-19 & Ebola genome sequences: 10233.39999999796 (34.22%)
COVID-19 & Camel_CoV genome sequences: 15134.599999998123 (50.61%)
COVID-19 & Malaria genome sequences: 8.800000000105774 (0.03%)
COVID-19 & HIV genome sequences: 5962.5999999990445 (19.94%)
COVID-19 & Hedgehog_CoV genome sequences: 15227.29999999811 (50.92%)
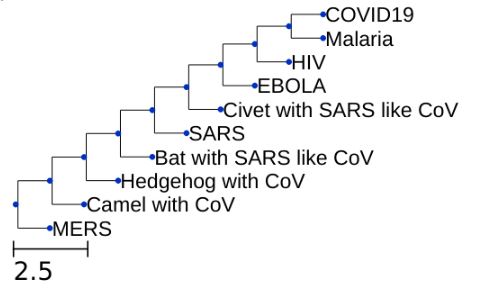
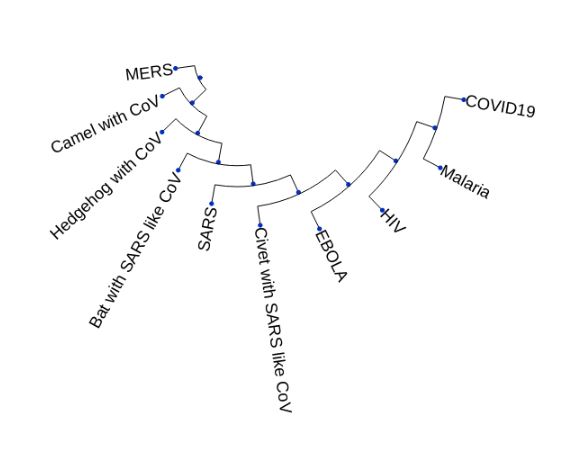
Generating a Phylogenetic Tree
A Phylogenetic Tree is the tree of evolutionary relationship among various species based on their genetic characteristics.
UPGMA (Unweighted Pair Group Method with Arithmetic mean) algorithm
This is where you can learn more about it wiki
[NOTE] unlike the above sequence similarity, here only the mismatch cases are scored using the aligner and no scoring is done for gaps and extended gaps.
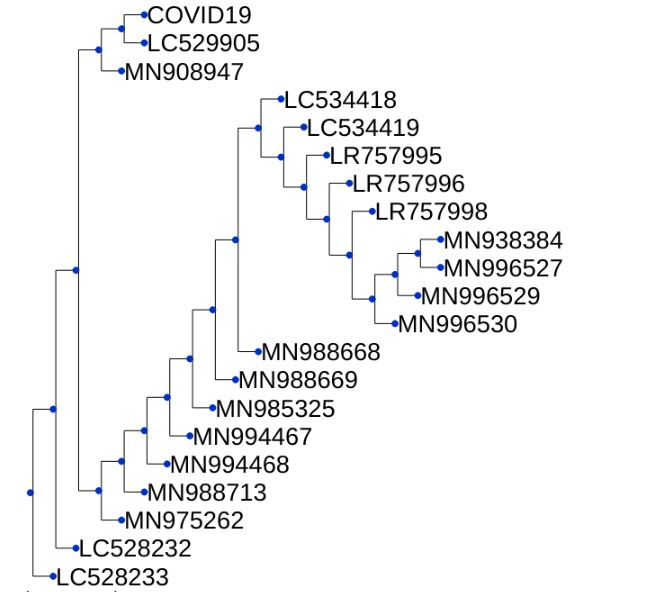
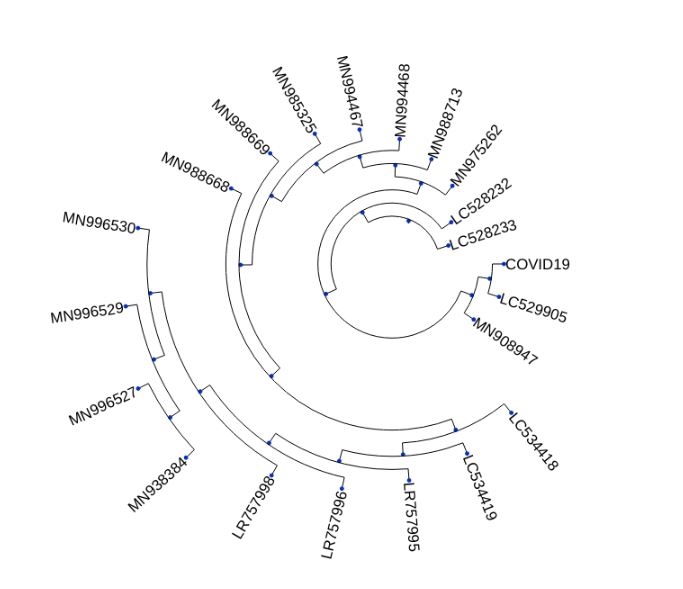
Clustering all COVID-19 patients’ genomes
using the UPGMA algorithm
Conclusions
- Based on the oligonucleotide compositions, all the genomes from coronaviridae family show similar composition (trinucleotide and tetranucleotide composition)
- Based on GC content, the composition of Hedgehog infected Corona virus is closely similar to COVID-19 genome’s GC content.
- Judging by the number of protein strands, the count in COVID19’s genome is very close to those found in Bat infected with SARS like corona virus.
- If you filter down some of the categories in the plotly-plot of distribution of amino acids, you’ll see the percentage of amino acids in COVID-19’s genome closely match to those of MERS, Civet infected with SARS like CoV, Camel with alpha CoV and Malaria except for some and low high contents among L (Leucine), P (Proline) and T (Threonine) amino acids.
- Checking by the generated Phylogenetic tree, the genomes of COVID-19 and malaria virus show the maximum similarity. Perhaps this also explains why there’s a lot of buzz on the news and internet about trying Hydroxycholorquine and anti-retroviral drugs which happen to be the medicines for malaria and HIV viruses, respectively.
- Of all the genomes of COVID-19 infected patients avaialable on Kaggle, some patients show similar traits when clustered based on their genome sequence similarity.I really don’t know how helpful this would be but if something works or had worked for a patient, then same should also work for patients in the same cluster.
If you liked this project, please consider buying me a coffee

Comments